Descargado 75 veces

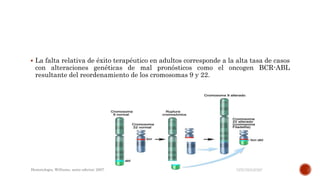




















El documento resume las características de la leucemia linfoblástica aguda (LLA), incluyendo que se origina de una única célula progenitora B o T, causa supresión de la hematopoyesis debido a la proliferación de blastos en la médula ósea, y puede acumularse extramedularmente. También discute factores de riesgo genéticos y ambientales, síntomas, hallazgos en exámenes de laboratorio e inmunofenotipado para el diagnóstico.























































































