Descargado 67 veces













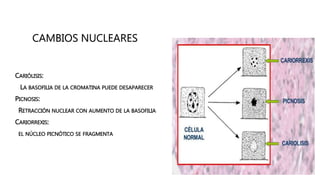



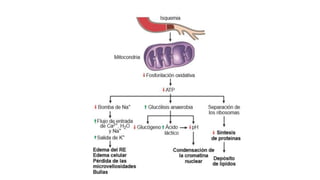

























La patología estudia los cambios estructurales y funcionales en células y tejidos debido a enfermedades, abordando aspectos como la etiología, patogenia y respuestas adaptativas. Se diferencian procesos como la necrosis y apoptosis, así como alteraciones celulares, mecanismos de lesión, y fenómenos como la calcificación patológica y el envejecimiento celular. Este análisis es esencial para entender cómo las células responden a estímulos nocivos y los mecanismos que llevan a la muerte celular.











































































































