Descargado 41 veces





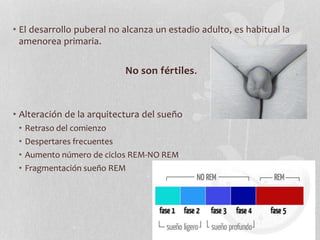


















El síndrome de Prader-Willi se caracteriza por hipotonía neonatal, problemas de alimentación en lactantes, obesidad en la niñez y rasgos faciales distintivos. Se debe a la falta de expresión de genes en la región 15q11-q13 del cromosoma 15 paterno. El diagnóstico se basa en criterios clínicos y genéticos como la detección de microdeleción en esa región. El tratamiento incluye dieta, fisioterapia y manejo de problemas asociados como déficit de GH o trastornos endocr























![Neurofibromatosis[1]](https://cdn.slidesharecdn.com/ss_thumbnails/neurofibromatosis1-121022043828-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



















































































